Determining the cause of difficult-to-control mitochondrial diseases
A Japanese research group has discovered that taurine is conjugated with mitochondrial transfer RNAs (tRNAs), small RNAs that decode genetic information, and that taurine deficiency in tRNA dramatically reduces both mitochondrial protein translation and impairs mitochondrial membrane integrity. Subsequently, many mitochondrial proteins are unable to localize on mitochondria and end up forming toxic aggregates inside the cell. Furthermore, researchers also found that some symptoms of mitochondrial dysfunction can be improved by maintaining protein quality.Mitochondria are intracellular organelles that function as the power plants of eukaryotic cells. Proteins gathered in mitochondria work on maintaining various functions. Thirteen species of mitochondrial proteins are encoded by mitochondrial DNA and produced in the mitochondria itself, while thousands of other mitochondrial proteins are produced in the cytosol and transported into the mitochondria.
When mutations occur in genes that design mitochondrial proteins, a severe neurological disorder called mitochondrial disease can develop. Mitochondrial myopathy, encephalomyopathy, lactic acidosis, stroke-like symptoms (MELAS) or Myoclonic Epilepsy with Ragged Red Fibers (MERRF) are major forms of mitochondrial disease that are characterized by whole body muscle weakness and declining cardiac function. To date, there is no effective treatment and many patients die a few years after onset.
The molecular mechanism underlying the pathogenesis of MELAS and MERRF is not yet fully understood, but it has been reported that taurine, one of the functional amino acids, is involved. In mitochondria, it is known that taurine binds to tRNA, which is also known to be related to protein synthesis, and that this binding is reduced in patients with MELAS and MERFF. However, the detailed molecular mechanisms by which the decrease of taurine binding to tRNA induces these serious symptoms was unknown.
To elucidate this mystery, the research group first identified the enzyme that conjugates taurine to the tRNA molecule. They then deleted the enzyme in a mouse model and found that protein translation in the mitochondria was greatly reduced. This result demonstrated that taurine is essential for protein synthesis in mitochondria.
Proteins produced in the mitochondria also serve to maintain mitochondrial structure. When examining mitochondria that had decreased taurine function, the researchers discovered that the inner mitochondria membrane had collapsed. Consequently, the various proteins produced outside the mitochondria, which are usually transported inside, could not enter the mitochondria. With nowhere to go, these protein structures eventually broke down, accumulated in the cytoplasm, and became highly toxic aggregates. The researchers believe this to be one of the causes of cytotoxicity that occurs in patients with MELAS and MERRF.
In recent years, compounds that inhibit abnormal protein aggregation have been developed. One of these compounds, TUDCA (ursodeoxycholic acid), is a substance originally present as a secondary bile acid in the body. Administering TUDCA to cells without taurine-tRNA binding resulted in the near complete disappearance of aggregates and the reduction of cellular stress. Moreover, when TUDCA was administered to mitochondrial disease model mice with reduced taurine-tRNA binding, cell damage was also improved.
"We believe that this research will lead to the development of therapeutic drugs for mitochondrial diseases in which taurine's activity declines, such as MELAS and MERRF," said Associate Professor Wei Fan-Yan of Kumamoto University, leader of the research project. "TUDCA, which has been shown to be effective in cell and model animals, is used as a remedy for liver diseases in Italy and other European countries, so its pharmaceutical safety has already been confirmed. We are planning to conduct clinical trials in the near future to determine whether TUDCA is an effective remedy for mitochondrial diseases in humans."
This research result was posted online in the journal "Cell Reports" on 9 January 2018.
[Source]
Fakruddin, M., Wei, F.-Y., Suzuki, T., Asano, K., Kaieda, T., Omori, A., … Tomizawa, K. (2018). Defective Mitochondrial tRNA Taurine Modification Activates Global Proteostress and Leads to Mitochondrial Disease. Cell Reports, 22(2), 482–496. doi:10.1016/j.celrep.2017.12.051
[Paper Info]
TITLE:
Defective mitochondrial tRNA taurine-modification activates global proteostress and leads to mitochondrial disease
AUTHORS:
Md Fakruddin, Fan-Yan Wei, Takeo Suzuki, Kana Asano, Takashi Kaieda, Akiko Omori, Ryoma Izumi, Atsushi Fujimura, Taku Kaitsuka, Keishi Miyata, Kimi Araki, Yuichi Oike, Luca Scorrano, Tsutomu Suzuki, and Kazuhito Tomizawa
JOURNAL: Cell Reports
DOI: 10.1016/j.celrep.2017.12.051
URL:
https://www.sciencedirect.com/science/article/pii/S2211124717318752?via%3Dihub
[Fund]
PRESTO (Precursory Research for Embryonic Science and Technology) from JST: "Creation of Innovative Technology for Medical Applications Based on the Global Analyses and Regulation of Disease-Related Metabolites"
http://www.jst.go.jp/kisoken/presto/en/research_area/ongoing/106sikkan.html
[Image]
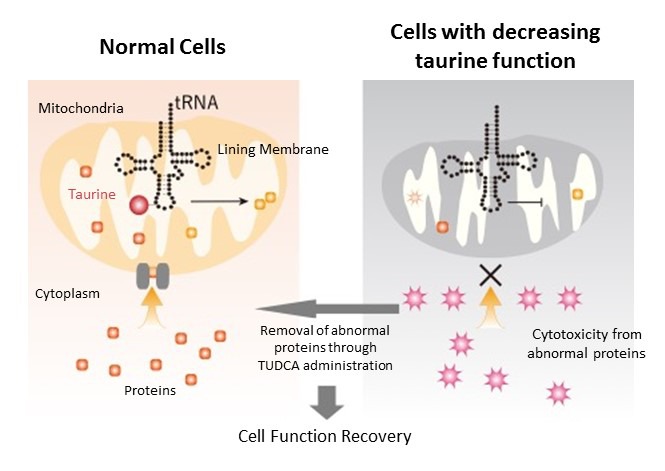
Protein production mechanism in mitochondria
CAPTION:
In mitochondria of normal cells, protein is synthesized by taurine binding with tRNA, and many proteins are transported from the cytoplasm to mitochondria for normal functionality. However, when taurine's activity declines, as in patients with MELAS or MERRF, protein synthesis in the mitochondria stops and the mitochondrial structure collapses. Proteins are then unable to enter the mitochondria from the cytoplasm so they break down and become aggregates that cause cell damage. When TUDCA is administered, abnormal protein accumulation is suppressed and cell function is restored.